The clearest signal yet that epilepsy treatment may finally be moving beyond symptom suppression arrived this month as Stoke Therapeutics and its partner Biogen released four-year open-label extension data for zorevunersen in Dravet syndrome, showing median seizure reductions of up to 91% sustained across years of treatment.
Dravet syndrome is a rare, severe genetic epileptic encephalopathy caused in most patients by loss-of-function variants in SCN1A, the gene encoding the voltage-gated sodium channel Nav1.1. Current standard-of-care options — including clobazam, valproate, and the recently approved fenfluramine and cannabidiol formulations — reduce seizure burden but do not address the underlying haploinsufficiency. Zorevunersen takes a different approach entirely.
The drug is an antisense oligonucleotide designed to bind unproductive SCN1A mRNA transcripts — those that would ordinarily be degraded through nonsense-mediated decay — and redirect them toward productive translation, increasing Nav1.1 protein expression in inhibitory interneurons. The mechanism targets the root cause of GABAergic dysfunction in Dravet rather than compensating downstream.
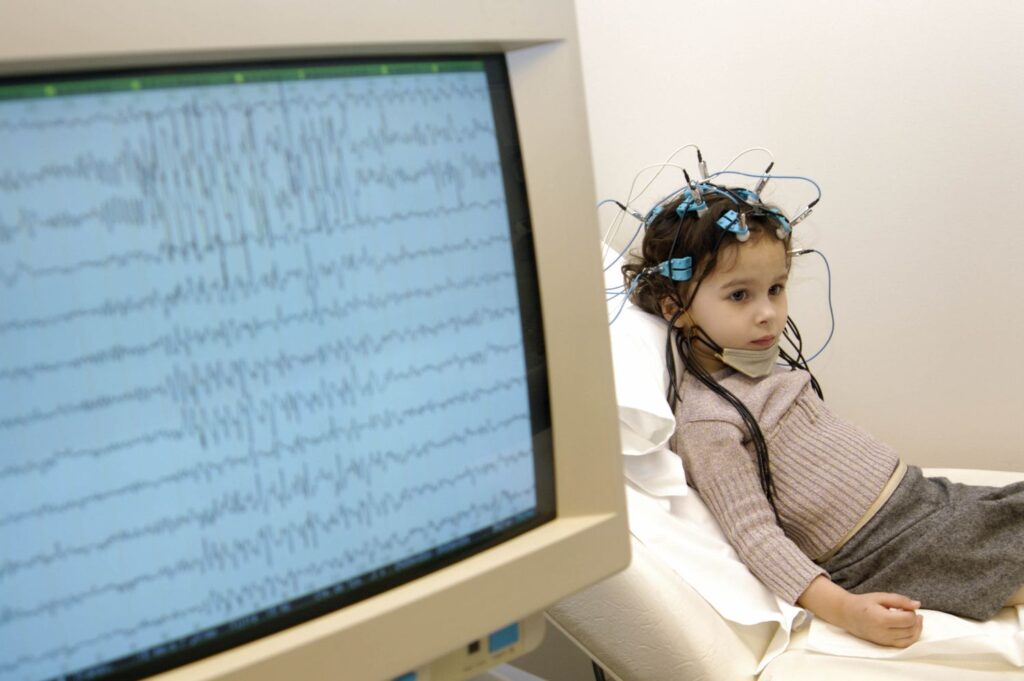
Data published in the New England Journal of Medicine in March 2026 from the MONARCH Phase 2/3 study demonstrated statistically significant reductions in monthly convulsive seizure frequency versus placebo over a 24-week treatment period. The extension cohort, now followed for up to 48 months, shows those gains not only persisting but deepening in a meaningful proportion of patients — with several children achieving near-seizure-free status for the first time in their lives.
Perhaps more clinically significant than the seizure numbers is what the long-term data suggest about neurodevelopmental trajectories. Caregivers and investigators report improvements in cognition, language acquisition, and adaptive behaviour in patients who have been on therapy the longest — effects that sodium channel modulators have never demonstrated. Whether these gains are attributable to reduced seizure burden or a more direct Nav1.1-mediated effect on synaptic inhibition remains an open scientific question, but one that is now being tracked prospectively.
The Phase 3 EMPEROR study, a randomised double-blind placebo-controlled trial enrolling 150 patients, was expected to complete enrolment by the end of Q2 2026, with a primary data readout anticipated in mid-2027. Stoke and Biogen have signalled intent to begin a rolling New Drug Application submission to the FDA in the first half of 2027, supported by the drug’s existing Breakthrough Therapy Designation.
The broader implications for the epilepsy field are significant. Zorevunersen’s progress validates the ASO platform for CNS indications characterised by monogenic aetiology — a category that extends well beyond Dravet to include CDKL5 deficiency disorder, KCNQ2 neonatal epilepsy, and a growing list of rare epileptic encephalopathies where the causative gene is known but no targeted therapy exists.
For drug developers working in adjacent mechanistic space — including those targeting the upstream glutamate-GABA balance that governs inhibitory tone more broadly — zorevunersen’s trajectory offers a proof point that durable disease modification in genetic epilepsy is an achievable clinical endpoint, not merely an aspiration.